
Sitemap
A list of all the posts and pages found on the site. For you robots out there is an XML version available for digesting as well.
Pages
Posts
portfolio
Atlantic Ocean
Published:
Perth, Australia

South China Sea
Published:
Hainan, China

publications
The chromatin-remodeling enzyme Smarca5 regulates erythrocyte aggregation via Keap1-Nrf2 signaling.
Published in eLife, 2021
Recommended citation: Ding, Y.*, Li, Y.*, Zhao, Z.*, Zhang, Q.C., and Liu, F. The chromatin-remodeling enzyme Smarca5 regulates erythrocyte aggregation via Keap1-Nrf2 signaling. eLife 10, e72557 (2021). https://elifesciences.org/articles/72557
CD127 imprints functional heterogeneity to diversify monocyte responses in inflammatory diseases.
Published in Journal of Experimental Medicine, 2022
Recommended citation: Zhang, B. et al. CD127 imprints functional heterogeneity to diversify monocyte responses in inflammatory diseases. Journal of Experimental Medicine 219, e20211191 (2022). https://rupress.org/jem/article/219/2/e20211191/212953/CD127-imprints-functional-heterogeneity-to
Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space.
Published in Nature communications , 2022
Recommended citation: Xiong, L.*, Tian, K.*, Li, Y., Ning, W., Gao, X., and Zhang, Q.C. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nat Commun 13, 1–17 (2022). https://www.nature.com/articles/s41467-022-33758-z
Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding.
Published in Cell Systems, 2024
Recommended citation: Li, Y.*, Zhang J.*, Gao, X., and Zhang, Q.C. Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding. Cell Syst (2024) May 29: S2405-4712(24)00124-8. https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00124-8
talks
Poster on Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding.
Published:
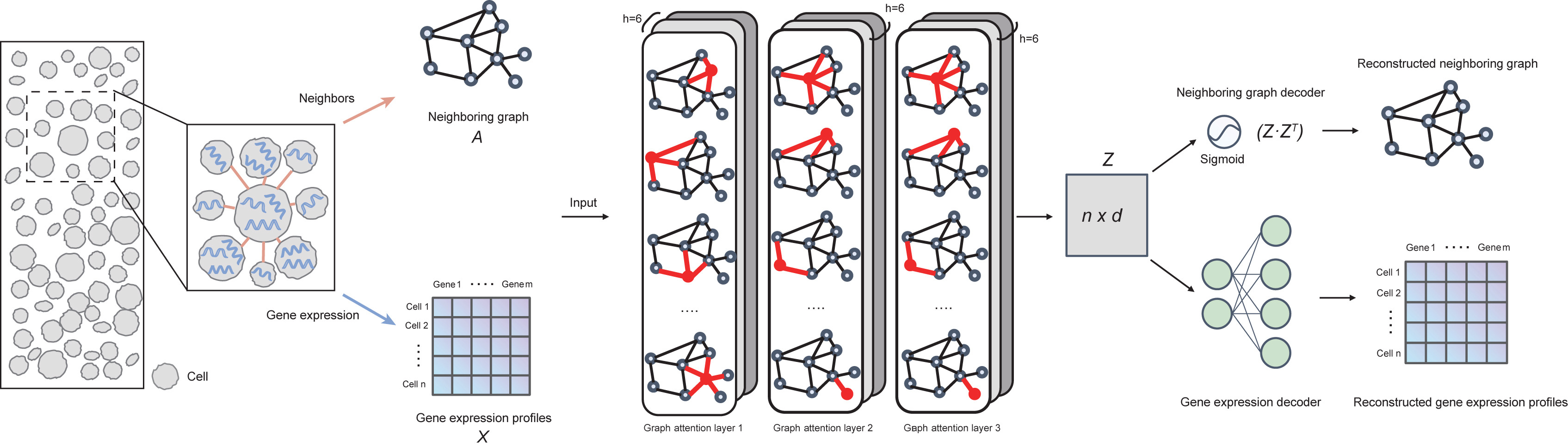
Computational methods are desired for single-cell-resolution spatial transcriptomics (ST) data analysis to uncover spatial organization principles for how individual cells exert tissue-specific functions. Here, we present ST data analysis via interaction-aware cell embedding (SPACE), a deep-learning method for cell-type identification and tissue module discovery from single-cell-resolution ST data by learning a cell representation that captures its gene expression profile and interactions with its spatial neighbors. SPACE identified spatially informed cell subtypes defined by their special spatial distribution patterns and distinct proximal-interacting cell types. SPACE also automatically discovered “cell communities”—tissue modules with discernible boundaries and a uniform spatial distribution of constituent cell types. For each cell community, SPACE outputs a characteristic proximal cell-cell interaction network associated with physiological processes, which can be used to refine ligand-receptor-based intercellular signaling analyses. We envision that SPACE can be used in large-scale ST projects to understand how proximal cell-cell interactions contribute to emergent biological functions within cell communities.
Poster on Single-Cell data Analysis via Perturbation Estimation
Published:
RNA velocity provides the directional information of cell-state dynamics and transitions from single-cell RNA-sequencing data. To reveal cellular transitions and perturb this process in silico, models capable of unveiling the causal effects of complex gene regulatory interactions are required. Here, we present SCAPE, a deep learning-based model that predicts the transcription dynamics under in silico genetic perturbations. SCAPE is uniquely able to predict in silico post-perturbation RNA velocities, which can be directly used to predict cell-fate diversions induced by the genetic perturbation of certain genes. We applied SCAPE to predict cell-fate diversions after in silico genetic perturbations in neurogenesis, gastrulation, and pancreatic endocrinogenesis. We validated the efficacy and accuracy of SCAPE in recapitulating well-characterized cell-state transitions induced by gene perturbations. Overall, SCAPE will facilitate the biomedical study of cell-fate decisions and cell-state transitions.
Poster on Single cell Causal Analysis of cell fate transition via in silico genetic PErturbation (SCAPE).
Published:
Dissecting the causal effects of complex gene regulatory interactions is essential for predicting cell fate, particularly during transitions induced by genetic perturbations. In this study, we introduce SCAPE, a deep learning-based method for modeling transcription dynamics that predicts the post-perturbation transcriptomic velocity field. This allows SCAPE to effectively predict cell fate changes resulted from genetic alterations in specific genes. We validated SCAPE’s performance using both simulated and real datasets, including those related to neurogenesis, gastrulation, pancreatic endocrinogenesis, endothelial-to-hematopoietic transition, and spermiogenesis. SCAPE demonstrates superior performance compared to competing methods in accurately recapitulating well-characterized cell state transitions induced by genetic perturbations. We anticipate that SCAPE will enhance our understanding of cell fate transitions and facilitate the discovery of key regulators in developmental processes and diseases.
teaching
Bioinformatics and Systems Biology Teaching Assistant
Graduate course, Prof. Qiangfeng Cliff Zhang, Tsinghua University, School of Life Sciences, 2021
This courses introduces the basics of bioinformatics, omics analysis, structural bioinformatics, systems biology, and the frontiers of bioinformatics and systems biology.
Bioinformatics and Systems Biology Teaching Assistant
Graduate course, Prof. Qiangfeng Cliff Zhang, Tsinghua University, School of Life Sciences, 2022
This courses introduces the basics of bioinformatics, omics analysis, structural bioinformatics, systems biology, and the frontiers of bioinformatics and systems biology.